genomics_applications
BIOINF 7150 Hi-C data analysis practical
Before we start, we need to download some files.
login to your vm.
cd
mkdir utils
cd utils
wget https://s3.amazonaws.com/hicfiles.tc4ga.com/public/juicer/juicer_tools_1.19.02.jar
wget https://raw.githubusercontent.com/nservant/HiC-Pro/master/bin/utils/hicpro2fithic.py
wget https://raw.githubusercontent.com/nservant/HiC-Pro/master/bin/utils/hicpro2juicebox.sh
cd
wget https://zerkalo.curie.fr/partage/HiC-Pro/HiCPro_testdata.tar.gz
wget http://zerkalo.curie.fr/partage/HiC-Pro/singularity_images/hicpro_2.11.3_ubuntu.img
cd miniconda3/envs/hic/lib/python3.6/site-packages/fithic
rm fithic.py
wget https://raw.githubusercontent.com/ningbioinfostruggling/fithic/master/fithic/fithic.py
cd
In this practical, we will learn the standard procedure of handling Hi-C data by processing them from raw sequencing data to statistically significant interactions.
First of all, watch this video to familiarize with the biological background of 3D genome: how does DNA loops are formed: https://www.youtube.com/watch?v=dES-ozV65u4
So now you know what the nature of DNA folding.
This DNA folding mechanism can further lead to different biological events, for example, DNA loops are formed and regions that located far away from each other can have close proximity three dimensionally. More importantly, such event can affect gene regulation if the DNA loops bring distal enhancer and promoter into close proximity.
Nowadays, the most popular way to investigate the 3D genome is by analysing Hi-C data.
Hi-C stands for high resolution chromosome conformation capture assay, it was invented in 2009 (https://www.ncbi.nlm.nih.gov/pubmed/19815776). The procedure to prepare a Hi-C library will be covered in this week’s lecture. 
In this practical, we will learn a standard workflow to analyse Hi-C data from raw sequencing reads to significant interactions, see Figure 1. We will use one of the most popular pipeline called HiC-Pro (https://github.com/nservant/HiC-Pro) to analyse a small dataset from Rao et al. 2014. We’ll also learn how to use a tool called Juicebox to visualise the interaction matrix and use Fit-Hi-C2 to identify significant interactions.
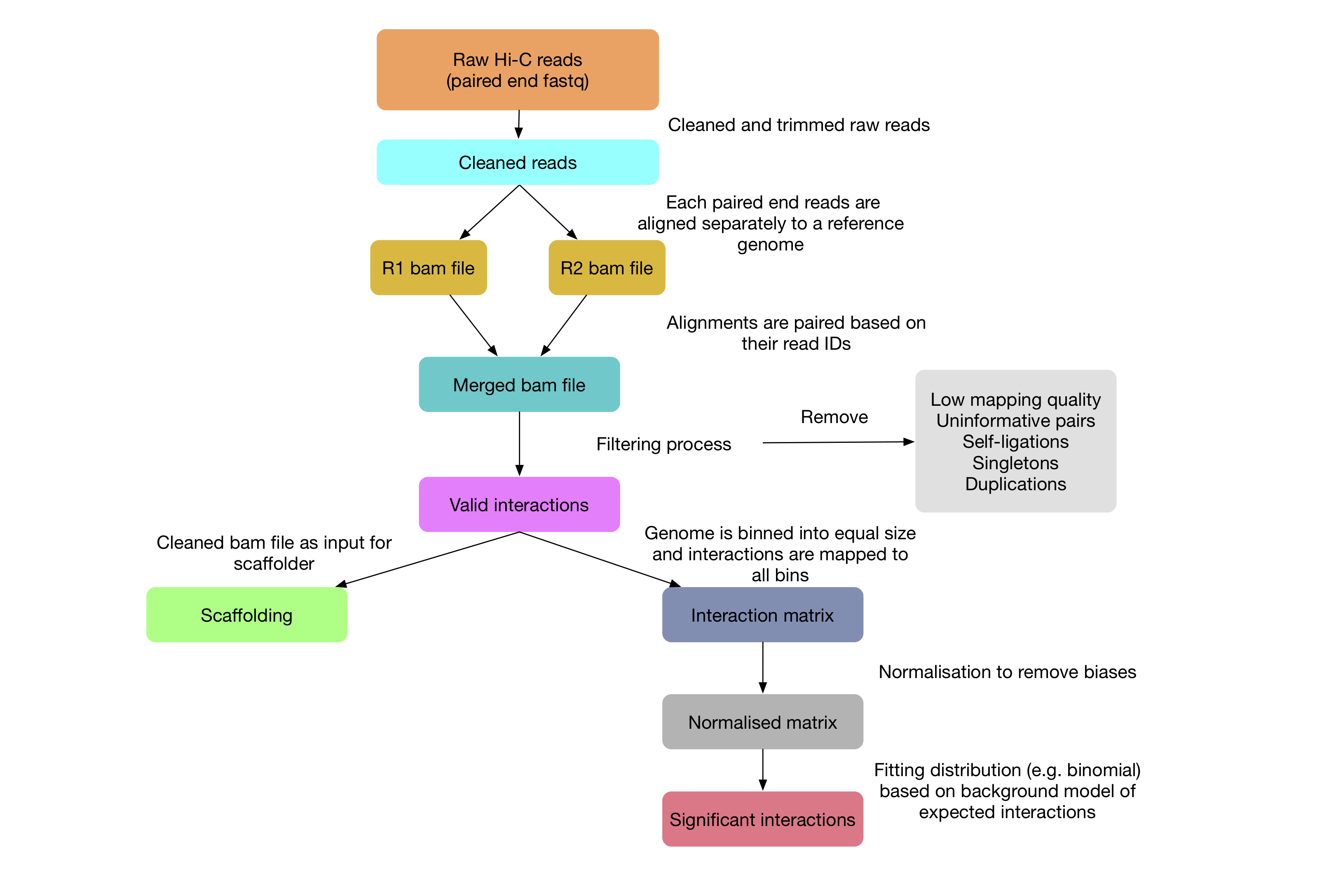 Figure 1: Standard workflow for analysing Hi-C data.
Figure 1: Standard workflow for analysing Hi-C data.
- in
~/, make sureHiCPro_testdata.tar.gzexists - make sure there is a
hg19-ref/, and doll hg19-ref/, check these files:
HindIII_resfrag_hg19.bedchrom_hg19.sizes- Bowtie2 index files of hg19 genomes that ends with
.bt2
-
hicpro_2.11.3_ubuntu.img -
a directory called
utilsand these files inside it:
hicpro2fithic.pyhicpro2juicebox.shjuicer_tools_1.19.02.jar
Check out HiC-Pro
Here we used the singularity container to use HiC-Pro.
To shell into the HiC-Pro image, do singularity shell hicpro_2.11.3_ubuntu.img
you should see Singularity hicpro_2.11.3_ubuntu.img:~>
Try HiC-Pro -h, see if you can see the help page of HiC-Pro.
Based on the help page, we can learn that there are three required parameters for running HiC-Pro, -i|--input INPUT, -o|--output OUTPUT and -c|--conf CONFIG.
Explain and edit the config file
The configure file is specific design for HiC-Pro to recognise all the parameters it need to run the pipeline.
First we get the configure file by doing:
tar -zxvf HiCPro_testdata.tar.gz
less config_test_latest.txt
Now we need to edit the configure file so HiC-Pro know where it can find all the files it need, including:
- reference genome
- genome size file
- restriction fragment file
Run HiC-Pro and explain every steps of it
In order to run HiC-Pro, first get into the singularity shell, but this time, we need files from local so we need to bind the files to it by doing:
singularity shell --bind /home/student:/mnt hicpro_2.11.3_ubuntu.img
And when we use the --bind parameter, we can find all the local files in /mnt, try ls -lh /mnt.
Now we can run HiC-Pro with the edited configure file.
HiC-Pro -c /mnt/config_test_latest.txt -i /mnt/test_data/ -o /mnt/test_out
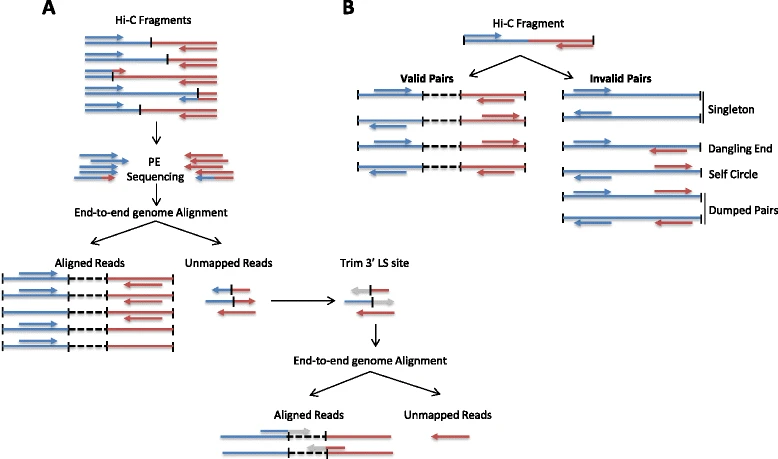
Inspect the results, answer these questions
-
What’s the uniquely mapping rates of of dixon_2M and dixon_2M_2?
-
Are all the uniquely mapping read pairs become valid interactions? If not, how many percentage of it becomes valid interactions?
-
What kinds of uninformative reads have been removed, please list at least three kinds? What’s the biggest contamination?
-
How many binned interactions have over 5 read pairs mapped to the bins at each resolution?
Identify significant interactions using FitHiC
- Why do we need to identify significant interactions?
Before we use FithiC, we need to check one thing by:
grep 'ningbioinfostruggling' miniconda3/envs/hic/lib/python3.6/site-packages/fithic/fithic.py
Source to hic environment and check out the help page of Fithic
source activate miniconda3/envs/hic/
fithic -h
To run Fithic, first we need to get the input for fithic, and it required python 2.7 while now we are in the environment of python 3
python2 utils/hicpro2fithic.py -h
This script can link the output of HiC-Pro to FitHiC
so we have 4 target to identify significant interactions, to avoid writing the command over and over again, we use loops:
mkdir fithic_input
cd fithic_input
sample=("dixon_2M" "dixon_2M_2")
res=("500000" 1000000)
for i in ${sample[@]}; do for j in ${res[@]}; do mkdir ${i}_${j}; python2 ../utils/hicpro2fithic.py -i ../test_out/hic_results/matrix/${i}/raw/${j}/*.matrix -b ../test_out/hic_results/matrix/${i}/raw/${j}/*bed -s ../test_out/hic_results/matrix/${i}/iced/${j}/*biases -o ${i}_${j} -r ${j}; done; done
Now we can again source to hic environment.
for j in ${res[@]}; do for i in dixon*${j}/; do name=$(basename $i); fithic -i ${i}/fithic.interactionCounts.gz -f ${i}/fithic.fragmentMappability.gz -r ${j} -t ${i}/fithic.biases.gz -o ./ -l ${name}; done; done
- At resolution of 500kb, how many significant interactions are identified with p-value <= 0.05?
- Think about what we can use with this interactions, i.e. how to give them biological meaning?
Visualise HiC interactions using heatmaps, introduce TADs and loops
Lastly we’ll try to visualise the interaction matrix.
Take dixon_2M at 500000 as example.
Like FitHiC, first we need to make the output of HiC-Pro compatible.
cd
mkdir juicebox_input
bash utils/hicpro2juicebox.sh -h
bash utils/hicpro2juicebox.sh -i test_out/hic_results/data/dixon_2M/dixon_2M.allValidPairs -g hg19-ref/chrom_hg19.sizes -j utils/juicer_tools_1.19.02.jar -r hg19-ref/HindIII_resfrag_hg19.bed -o juicebox_input/
Show the Juicebox web-browser version at https://aidenlab.org/juicebox/