Quantify and Filter putative double strand DNA from strand-specific RNA bam file
strandCheckR-package.RdThis package aims to quantify and remove putative double strand DNA from a strand-specific RNA sample. There are also options and methods to plot the positive/negative proportions of all sliding windows, which allow users to have an idea of how much the sample was contaminated and the appropriate threshold to be used for filtering.
Details
The package has some following main functions:
- getStrandFromBamFile: calculate positive/negative proprortion and
sum of reads over all sliding windows from a bam file
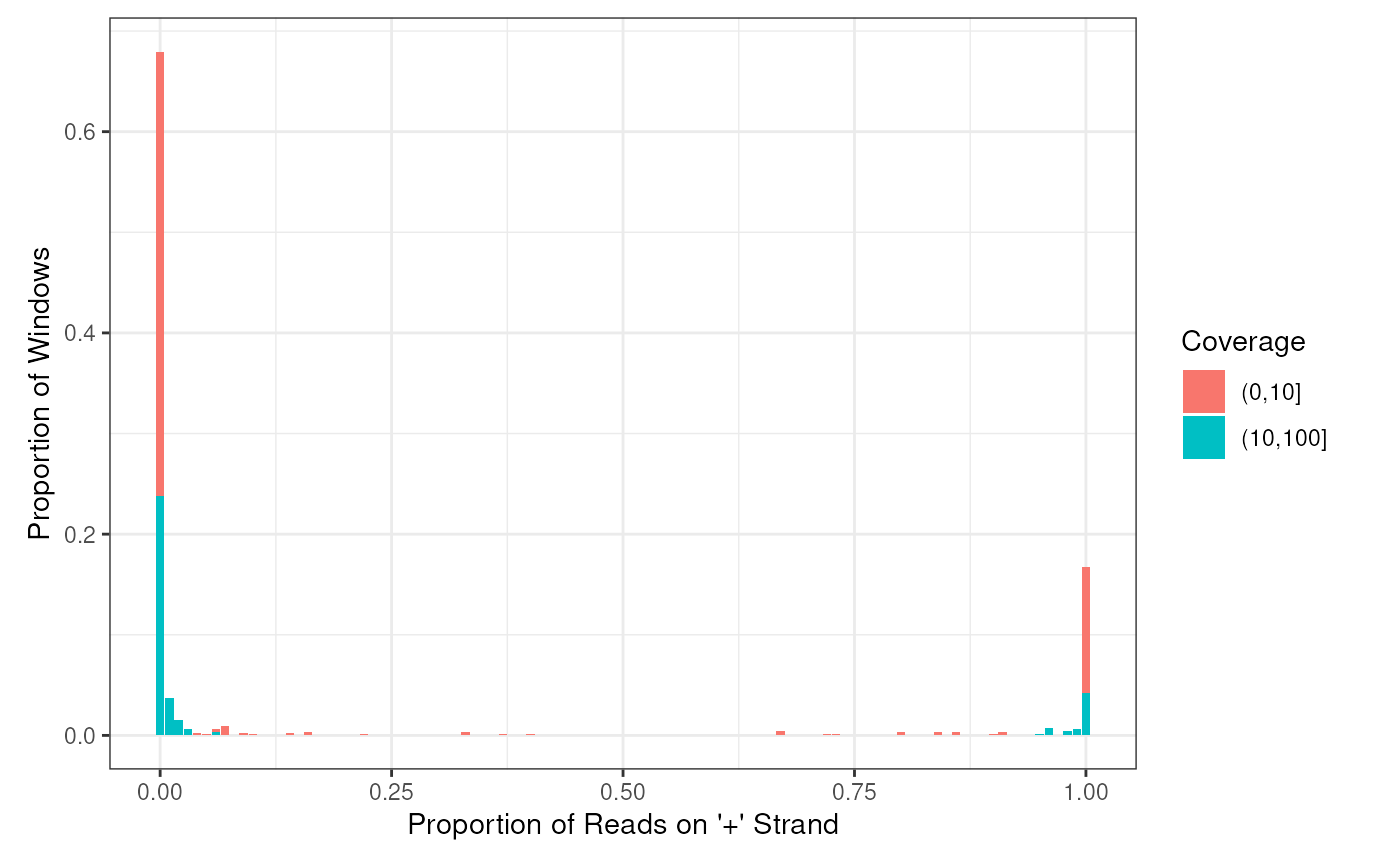
- plotHist: plot histogram of positive proportion of windows
calculated from getStrandFromBamFile method
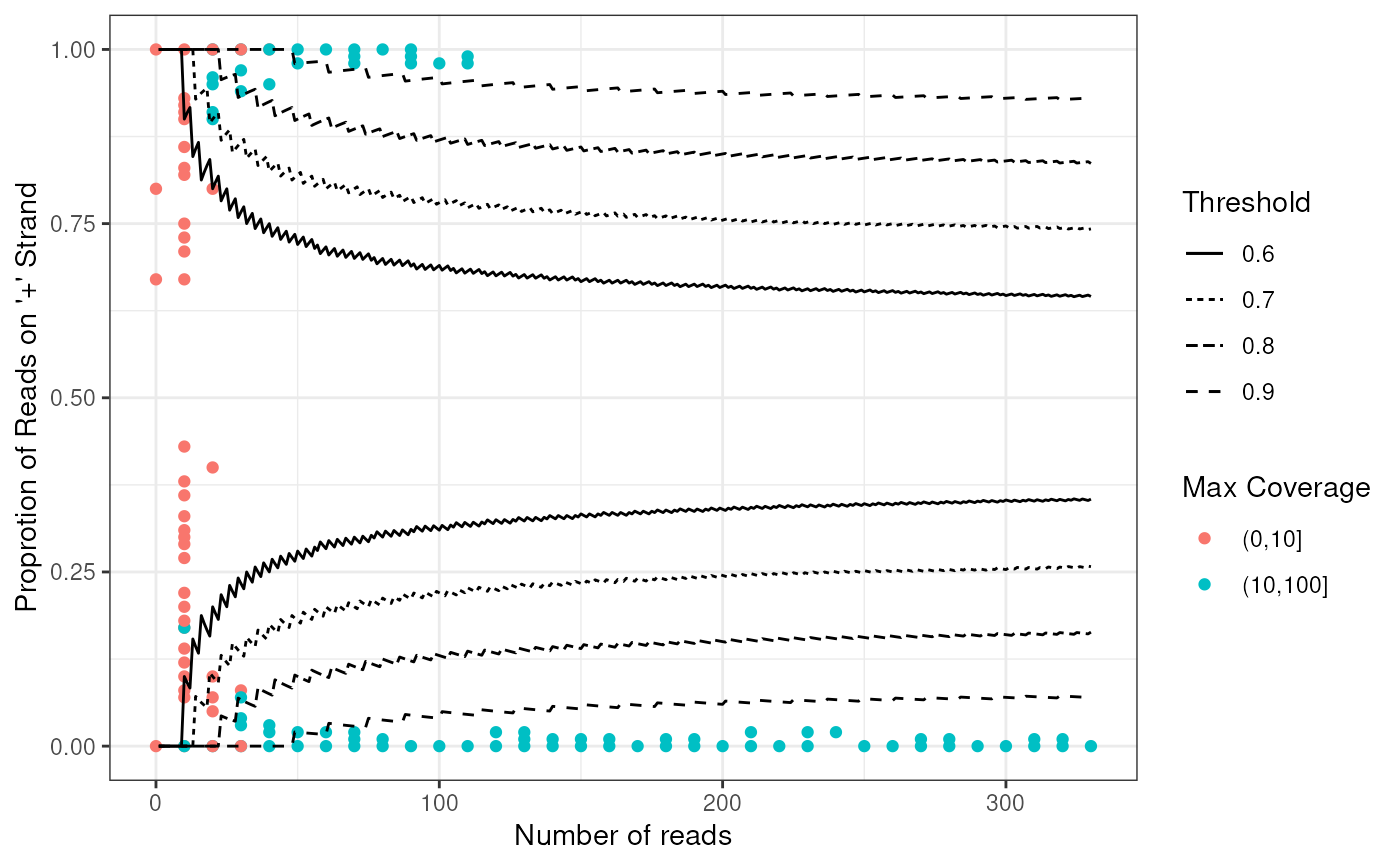
- plotWin: plot positive proportion vs number of reads of
windows calculated from getStrandFromBamFile method
- filterDNA: filter a bam file
Examples
bamfilein <- system.file("extdata","s1.sorted.bam",package = "strandCheckR")
windows <- getStrandFromBamFile(bamfilein)
#> Testing paired end by checking the first 1e+05 reads of file /__w/_temp/Library/strandCheckR/extdata/s1.sorted.bam
#> Your bam file is single end
#> Reading file /__w/_temp/Library/strandCheckR/extdata/s1.sorted.bam
#> Read sequences 10
plotWin(windows)
#> Warning: Removed 1 row containing missing values or values outside the scale range
#> (`geom_point()`).
 plotHist(windows)
plotHist(windows)
 filterDNA(file = bamfilein,destination = "filter.bam")
#> Testing paired end by checking the first 1e+05 reads of file /__w/_temp/Library/strandCheckR/extdata/s1.sorted.bam
#> Your bam file is single end
#> Summary will be written to out.stat
#> Read sequences 10
filterDNA(file = bamfilein,destination = "filter.bam")
#> Testing paired end by checking the first 1e+05 reads of file /__w/_temp/Library/strandCheckR/extdata/s1.sorted.bam
#> Your bam file is single end
#> Summary will be written to out.stat
#> Read sequences 10